NEUROD2, un nouveau gène d’autisme identifié chez la souris !
Par criblage génétique les scientifiques ont isolé le gène Neurod2 comme enrichi dans les neurones au moment de leur intégration synaptique. Pour étudier son rôle ils ont caractérisé les souris déficientes pour Neurod2 à de multiples échelles, de la molécule au comportement. Ces analyses montrent un ensemble d’altérations typiques de l’autisme ou de modèles rongeurs confirmés d’autisme. Forts de ces découvertes, les chercheurs ont cherché et identifié 11 patients autistes avec mutations causatives dans NEUROD2.
Cette recherche se focalise sur les facteurs moléculaires qui régulent le développement du cerveau et plus particulièrement du cortex cérébral, zone la plus fortement associée à l’autisme. En utilisant un crible génétique chez la souris les scientifiques ont isolé Neurod2 comme facteur enrichi dans les neurones au moment de la maturation synaptique. Bien que le gène NEUROD2 ait récemment été associé à l’épilepsie chez deux patients, son rôle dans le cerveau demeure mal connu. Pour pallier à cela, ils ont étudié la fonction de Neurod2 par une approche de génétique inverse - qui consiste à étudier les altérations visibles chez une souris déficiente (KO) pour le gène Neurod2 -, de la molécule au comportement. Chez les embryons Neurod2 KO, les neurones corticaux migrent anormalement – ils dépassent leur zone cible prévue, s’arrêtant trop loin dans leur voyage. Cela altère l’organisation laminaire de la structure corticale malgré le fait que son épaisseur totale est normale. Chez les souris juvéniles et adultes, la densité et le remodelage des synapses sont dérégulés spécifiquement dans les prolongements apicaux des neurones corticaux, ce qui est une caractéristique de modèles déjà connus d’autismes avec autres origines génétiques. L’enregistrement de l’activité électrique des neurones corticaux a révélé une excitabilité intrinsèque anormalement élevée chez les souris Neurod2 KO comparée à leurs congénères sauvages. Au niveau moléculaire, un séquençage haut débit de cellules issues de cortex a montré une expression anormale de nombreux gènes connus comme ayant des rôles importants dans l'excitabilité neuronale et à la fonction synaptique. Les « cousins » humains de ces gènes exprimés à des niveaux anormaux se trouvent être pour beaucoup déjà connus comme fortement associés aux troubles autistiques. Au niveau comportemental enfin, les souris Neurod2 KO présentent des déficits d'interaction sociale, des stéréotypies, une hyperactivité et parfois des crises épileptiques spontanées. Les souris hétérozygotes pour Neurod2, c’est-à-dire disposant d’une copie de Neurod2 sur les 2 habituelles, présentaient des défauts similaires aux souris complètement déficientes. Ceci indique que Neurod2 est haploinsuffisant – que la perte d’une seule des 2 copies du gène est déjà délétère. Enfin, la suppression de Neurod2 spécifiquement dans les neurones excitateurs du cortex est suffisante pour récapituler les problèmes cellulaires et comportementaux trouvés chez les souris KO complètes (dans lesquelles Neurod2 est enlevé dans absolument toutes les cellules de l’organisme), révélant que c’est bien l’expression de Neurod2 dans le cortex qui génère les symptômes autistiques. Pour réaliser cette expérience, les scientifiques ont généré une nouvelle souche de souris « KO conditionnelle » dans laquelle Neurod2 peut être enlevée à l’envi dans l’espace et le temps en utilisant un tour de passe-passe génétique. Cette lignée sera très intéressante à l’avenir pour étudier la fonction précise de Neurod2 dans des sous-populations de neurones ou des zones cérébrales précises.
Informés par ces caractéristiques des souris mutantes KO complètes ou conditionnelles, les chercheurs ont sollicité des cliniciens et généticiens humains. Cela a permis d’identifier onze patients avec trouble autistique et déficience intellectuelle causativement associés à des mutations pathogènes dans NEUROD2. Pour chaque patient, la mutation ne concerne qu’une seule base d’ADN sur plus de 1000 (on parle de mutation ponctuelle), mais celle-ci suffit à faire perdre totalement sa fonction au produit protéique du gène NEUROD2 et donc équivaut à une perte du gène comme chez la souris.
Ces résultats démontrent le rôle crucial pour Neurod2 dans le développement néocortical, dont les altérations peuvent provoquer des troubles neurodéveloppementaux, notamment la déficience intellectuelle et les troubles du spectre de l'autisme (TSA).
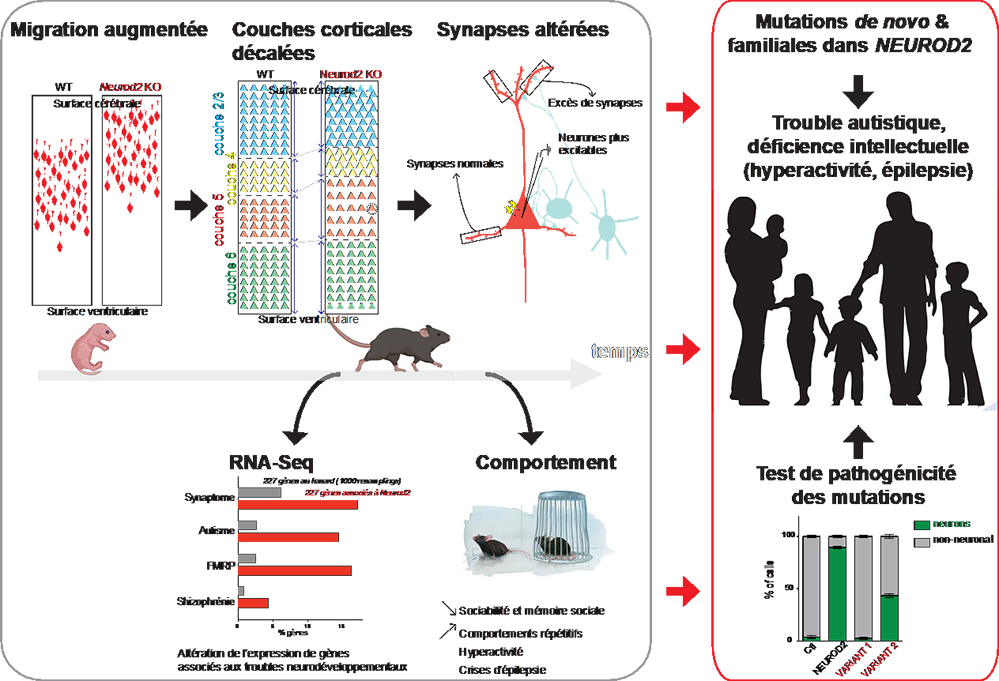
Figure : Encadré noir (de gauche à droite et de haut en bas): résumé des altérations anatomo-comportementales observées chez la souris dépourvue de Neurod2. Dans le cortex d’embryons, les jeunes neurones migrent anormalement loin vers la surface, ce qui induit un décalage superficiel des couches corticales chez la souris adolescente. On observe aussi un excès de synapses dans la partie « apicale » des neurones corticaux. Un séquençage par RNA-seq a montré un certain nombre de gènes (exactement 227 ayant des « cousins » humains) dont l’expression est altérée chez les souris dépourvues de Neurod2. Le graphe montre que les chevauchements entre cette liste de gènes régulés par Neurod2 et les listes de gènes connus associés aux synapses, à l’autisme, l'X Fragile et la schizophrénie sont très significativement supérieurs à ceux obtenus en prenant au hasard le même nombre de gènes dans le génome. Cela démontre que Neurod2 joue un rôle central dans les voies de signalisation associées à ces troubles. Enfin, les souris Neurod2 KO montrent tous les signes comportementaux de l’autisme.
Encadré rouge : Les scientifiques ont trouvé 11 patients présentant des mutations dans NEUROD2. Ces patients présentent tous un trouble autistique avec déficience intellectuelle, et certains présentent en plus des comorbidités telles que l’hyperactivité et l’épilepsie. Enfin, ils ont mis au point un test simple in vitro qui permet de démontrer la pathogénicité des variants NEUROD2 chez les patients.
Pour en savoir plus :
Disruption of NEUROD2 causes a neurodevelopmental syndrome with autistic features via cell-autonomous defects in forebrain glutamatergic neurons.
Runge K, Mathieu R, Bugeon S, Lafi S, Beurrier C, Sahu S, Schaller F, LoubatA, Herault L, Gaillard S, Pallesi-Pocachard E, Montheil A, Bosio A, Rosenfeld JA, Hudson E, Lindstrom K, Mercimek-Andrews S, Jeffries L, van Haeringen A, Vanakker O, Van Hecke A, Amrom D, Küry S, Ratner C, Jethva R, Gamble C, Jacq B, Fasano L, Santpere G, Lorente-Galdos B, Sestan N, Gelot A, Giacuzz S, Goebbels S, Represa A, Cardoso C, Cremer H, de Chevigny A.
Molecular Psychiatry. 2021 Jun 29. doi: 10.1038/s41380-021-01179-x.
Contact
laboratoire
Institut de neurobiologie de la méditerranée (INMED) (Inserm/Aix-Marseille University)
Parc Scientifique de Luminy
13273 MARSEILLE Cedex 09
FRANCE