Altérations très précoces des régulations épigénétiques dans la maladie de Huntington
La maladie de Huntington affecte de façon primaire les neurones du striatum, structure cérébrale impliquée dans le contrôle des mouvements volontaires et dans certaines fonctions cognitives. Dans cette étude parue dans la revue Nature Communications, les scientifiques montrent que, chez des souris modèles de la maladie, les mécanismes épigénétiques qui contribuent à l’intégrité des neurones du striatum sont altérés bien avant l’apparition des troubles moteurs. Ces nouvelles données apportent une preuve moléculaire de la détérioration précoce et progressive du striatum « Huntington ».
La maladie de Huntington est une maladie génétique rare, diagnostiquée généralement à l’âge adulte et entraînant la mort au terme d’une évolution de 10-15 ans. Elle se caractérise par des symptômes moteurs (des mouvements involontaires) et comportementaux typiques, résultant d’une atteinte primaire des neurones du striatum, une région sous-corticale du cerveau. Quels sont les mécanismes qui sous-tendent la mort (ou dégénérescence) des neurones striataux ? Quand ces mécanismes se mettent-il en place ? Ces questions restent encore largement ouvertes. Y apporter des réponses est essentiel, puisque le succès d’une thérapie dépend à la fois de la spécificité du traitement et de la précocité du diagnostic.
La dégénérescence des neurones du striatum chez les patients Huntington survient au terme d’une longue période de dysfonctionnement, portant atteinte à leur identité. Au cours du développement, les cellules se différencient en types cellulaires définis (par exemple, une cellule musculaire, hépatique, neuronale, etc…), acquérant ainsi une fonction et une identité précises. L’acquisition et le maintien d’une fonction/identité cellulaire au sein d’un organisme est sous le contrôle de mécanismes épigénétiques, c’est-à-dire de mécanismes capables de modifier l’état de compaction de la chromatine, et en conséquence l’accessibilité des gènes. Ainsi, certaines modifications chimiques sur les histones -un composant majeur de la chromatine- comme l’acétylation favorisent un état relâché de la chromatine, permettant l’expression des gènes. Les gènes qui définissent la fonction/l’identité d’une cellule sont fortement exprimés, et la chromatine qui les porte est régionalement hyper-acétylée.
On savait que l’expression des gènes de l’identité striatale et l’acétylation de la chromatine associée étaient réduites chez les patients Huntington et chez des souris modèles symptomatiques. Cette nouvelle étude révèle que la perte sélective d’acétylation des histones au niveau des gènes de l’identité striatale se met en place très précocement, bien avant l’apparition des déficits moteurs chez la souris, et que ce processus a pour effet une accélération des mécanismes épigénétiques liés au vieillissement.
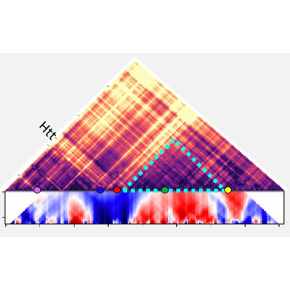
La maladie de Huntington est causée par une mutation unique, une répétition anormale de codons CAG dans le gène Huntingtine (HTT). Les scientifiques montrent que des altérations épigénétiques ciblent plus spécifiquement le gène muté. Les résultats indiquent que la présence de la mutation chez la souris modifie précocement l’organisation tridimensionnelle de la chromatine et l’expression des gènes associés.
Ces travaux améliorent ainsi la compréhension du processus pathogénique de la maladie de Huntington, en montrant que la mutation en cause affecte précocement la chromatine du striatum par deux mécanismes distincts.
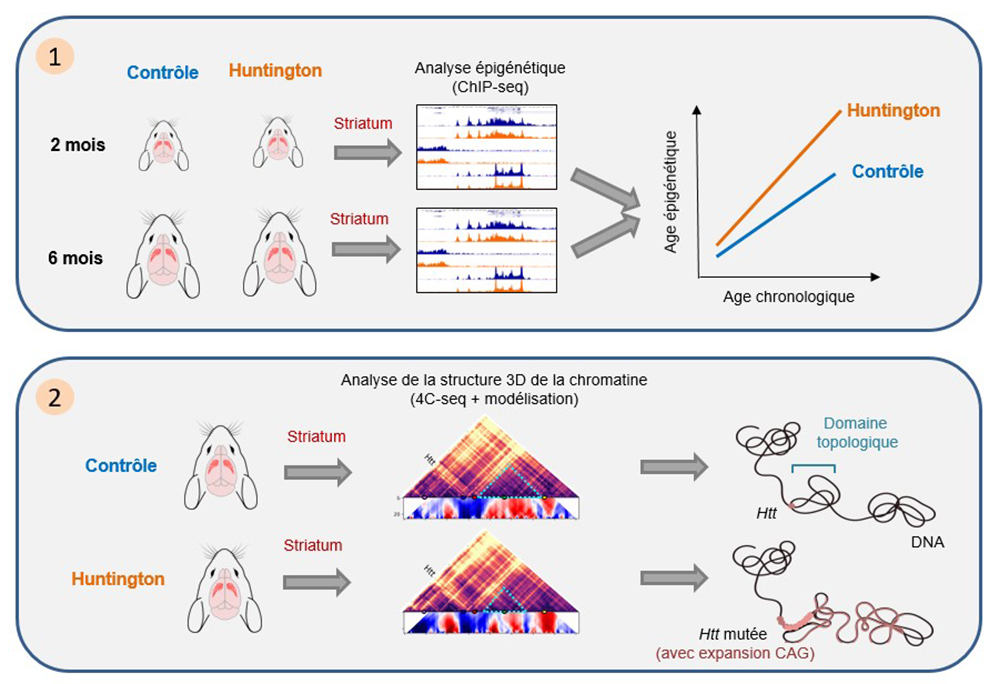
Figure :
1. L’analyse des profils épigénétiques par immunoprécipitation de la chromatine couplée à du séquençage haut-débit (ChIPseq) montre une accélération précoce des altérations épigénétiques associées au vieillissement dans le striatum de souris modèles de la maladie de Huntington.
2. L’analyse de la chromatine par la technique de "chromosome conformation capture" associée à du séquençage haut-débit (4C-seq) montre une ré-organisation de la structure spatiale de la chromatine au niveau du gène muté dans le striatum de souris modèles de la maladie de Huntington.
Pour en savoir plus :
Age-related and disease locus-specific mechanisms contribute to early remodelling of chromatin structure in Huntington’s disease mice
Alcal.-Vida R, Seguin J, Lotz C, Molitor AM, Irastorza-Azcarate I, Awada A, Karasu N, Bombardier A, Cosquer B, Gomez Skarmeta JL, Cassel JC, Boutillier AL, Sexton T, Merienne K
Nature communications 13 Janvier 2021. https://doi.org/10.1038/s41467-020-20605-2
Contact
Laboratoire
Laboratoire de neurosciences cognitives et adaptatives (LNCA) - (CNRS/Universite de Strasbourg)
12 rue Goethe, 67000 Strasbourg