Un senseur nutritionnel dans l’estomac de la cellule
Pour rester en bon état de marche, les cellules qui ne se divisent plus, comme les neurones, renouvellent leurs composants en les dégradant et en les recyclant dans des compartiments acides appelés lysosomes. Ces "micro-estomacs" servent aussi de senseurs et d’intégrateurs de signaux internes et externes pour coordonner les réponses cellulaires. Une étude publiée dans la revue PNAS montre comment les fonctions de recyclage et de senseur s’articulent au sein d’une même protéine dans la membrane du lysosome.
Nos cellules sont dotées de petites structures acides, appelées lysosomes, capables de digérer et recycler les composants cellulaires. Ceci permet d’éliminer les composants âgés ou défectueux et de recycler leurs briques élémentaires (acides aminés, sucres, etc…), un phénomène connu sous le nom d’autophagie. Cette opération de maintenance cellulaire est particulièrement importante dans les cellules à longue durée de vie, comme les neurones. C’est pourquoi une dysfonction des lysosomes induit un large spectre de maladies, depuis des maladies génétiques rares de surcharge lysosomale jusqu’à des maladies neurodégénératives fréquentes comme la maladie de Parkinson, où la mutation de gènes lysosomaux est un facteur de risque important.
Les lysosomes servent aussi de senseurs de nutriments et de plateformes de signalisation intracellulaire, ce qui permet de contrôler leur fonction dégradative, potentiellement dangereuse, et de la coordonner avec d’autres réponses cellulaires. L’articulation entre les fonctions de dégradation et de signalisation des lysosomes est complexe et mal comprise. Dans cette étude, les chercheurs ont étudié une protéine de la membrane lysosomale, baptisée PQLC2, qui conjugue ces deux fonctions. Elle sert à la fois de transporteur d’acides aminés basiques (lysine, histidine, arginine) issus de la dégradation lysosomale et de plateforme de recrutement d’un complexe protéique, appelé CSW, dont l’un des gènes, C9ORF72, est muté dans des formes familiales de sclérose latérale amyotrophique et de dégénérescences lobaires fronto-temporales.
Grâce à une approche électrophysiologique permettant de mesurer en temps réel l’activité de transport de PQLC2, les chercheurs ont observé que celle-ci est fortement modulée par le potentiel électrique de la membrane lysosomale, un paramètre reflétant l’état nutritionnel et énergétique de la cellule. Ces mesures ont aussi révélé que, parmi les substrats de PQLC2, l’arginine joue un rôle paradoxal car, selon les conditions, elle peut tout autant stimuler que freiner l’activité de transport. Des simulations numériques montrent que ce paradoxe peut s’expliquer par un effet spécifique de l’arginine sur une double transition structurale inhérente à l’activité de transport de PQLC2, que l’on peut comparer à un mécanisme de sas de sécurité à deux portes.
Mais, alors que le transport d’acides aminés par PQLC2 nécessite un cycle complet de cette transition, sa fonction de signalisation, c’est-à-dire son rôle de récepteur lysosomal du complexe CSW lors d’une carence en acides aminés, correspondrait à un arrêt dans une conformation particulière du cycle favorable au recrutement du complexe. La bascule de la fonction de transporteur vers celle de récepteur serait déclenchée par l’inversion de polarité électrique de la membrane lysosomale induite par la carence, décrite dans des travaux antérieurs. Dans un deuxième temps, le réapprovisionnement de la cellule en arginine grâce à la mise en route de l’autophagie écourterait ces pauses conformationnelles en raison de l’interaction particulière de l'arginine avec les « portes » de PQLC2, ce qui détacherait le complexe CSW des lysosomes pour interrompre sa signalisation. Par la suite, la résolution complète de la carence nutritionnelle par l’autophagie permettrait à la membrane lysosomale de retrouver sa polarité électrique initiale pour réactiver la fonction de transport de PQLC2.
Ce mécanisme montre comment une protéine membranaire peut servir simultanément de transporteur et de senseur/récepteur pour coordonner deux types de fonction d’un organite intracellulaire. Interférer avec ce mécanisme permettra de comprendre quel est le rôle du complexe CSW au lysosome, qui reste énigmatique, et d’évaluer son implication dans la pathogenèse de certaines formes de sclérose latérale amyotrophique et de dégénérescences fronto-temporales.
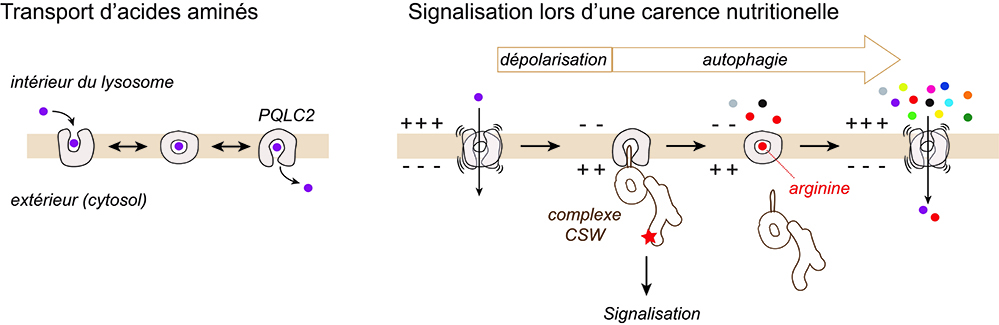
Figure : Dualité fonctionnelle de la protéine lysosomale PQLC2. En conditions normales, PQLC2 exporte des acides aminés du lysosome par un mécanisme analogue à un sas de sécurité à deux portes. Lors d’une carence nutritionnelle, l’inversion de polarité électrique du lysosome induit des pauses de PQLC2 dans une conformation ouverte vers le cytosol, favorable au recrutement du complexe de signalisation CSW. Simultanément, l’activation de l’autophagie restaure progressivement les stocks d’acides aminés. La liaison de l’arginine à PQLC2 stimule la refermeture de sa porte externe, ce qui décroche le complexe CSW et arrête sa signalisation. La résolution complète de la carence nutritionnelle par l’autophagie repolarise ensuite la membrane lysosomale.
Pour en savoir plus :
Arginine-selective modulation of the lysosomal transporter PQLC2 through a gate-tuning mechanism.
Xavier Leray, Rossella Conti, Yan Li, Cécile Debacker, Florence Castelli, François Fenaille, Anselm A. Zdebik, Michael Pusch & Bruno Gasnier
PNAS 10 aout 2021 . https://doi.org/10.1073/pnas.2025315118
Contact
laboratoire
Saints-Pères Paris Institute for the Neurosciences (SPPIN) (Université de Paris/CNRS)
45 rue des Saints-Pères
75006 Paris
France