
Chez la bactérie, l’activation de chaque gène induit la formation de mini domaines à l’intérieur des chromosomes
Dans un article publié dans la revue Nature Structural and Molecular Biology, ...
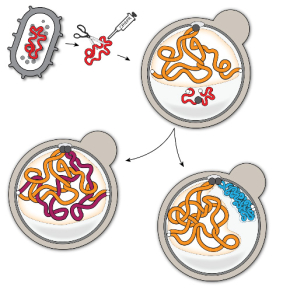
Les transferts d’ADN d’une espèce à une autre sont des événements fréquents et naturels. Mais suite à cette intégration, comment l’ADN s’adapte-t-il aux règles métaboliques de son nouvel hôte ? Dans un article publié dans Science, des scientifiques ont étudié des génomes bactériens entiers artificiellement insérés dans des cellules de levure, et ont montré que leur organisation et leur expression peuvent être prédites depuis leur séquence ADN grâce à des approches d’apprentissage profond. Ces résultats éclairent les processus évolutifs qui accompagnent les transferts d’éléments génétiques mobiles et ouvrent de nouvelles perspectives pour des applications biotechnologiques.
La composition de la séquence de l’ADN des génomes, c’est-à-dire la proportion relative des bases, notamment la guanine et la cytosine (appelée « contenu en GC »), ainsi que la présence de motifs répétés, varient d’une espèce à l’autre et au sein d’un même génome. Dans les cellules eucaryotes, qui possèdent un noyau, ces caractéristiques influencent directement l’expression des gènes et l’organisation tridimensionnelle des chromosomes. Le génome de chaque espèce évolue donc avec les complexes protéiques qu’il encode et qui contrôlent ces propriétés. Or, il arrive que de l'ADN étranger, comme des éléments mobiles exogènes, des virus, ou encore des segments d’ADN contenant des gènes, intègre ou envahisse naturellement ou artificiellement (i.e. en laboratoire) le noyau d’une cellule. Cet ADN est alors confronté à des mécanismes de régulation sous les règles desquelles il n’a pas évolué. La manière dont les cellules hôtes finissent par adopter ces nouvelles séquences exogènes reste largement inexplorée. Seront-elles activement transcrites ou non ? Certains facteurs favoriseront-ils leur adoption ou, au contraire, leur élimination ? Répondre à ces questions permettrait de mieux comprendre les mécanismes moléculaires régissant l’adaptation de séquences exogènes d’ADN à l'environnement d’un génome hôte, et d’apporter un nouvel éclairage à des phénomènes liés à l’évolution, au transfert de gènes entre espèces ou encore pour optimiser les approches de modification génétique en biotechnologie.
Pour répondre à ces questions, des scientifiques, dans un travail publié dans Science, ont étudié des molécules d’ADN exogènes de grande taille et présentant différentes compositions intégrées dans le génome de la levure Saccharomyces cerevisiae. Ces ADN exogènes pouvaient ainsi être des génomes bactériens entiers, de l’ordre de 1 mégabase (représentant environ 7% de la taille du génome de l’hôte), ou encore des séquences provenant du génome d’un autre eucaryote, Plasmodium falciparum. À chaque fois, la composition, l’activité transcriptionnelle, ainsi que le repliement de ces séquences dans le noyau au cours du cycle cellulaire ont été étudiés ainsi que les mécanismes moléculaires qui régissent leur adaptation à l’environnement de la cellule hôte. Enfin, les chercheurs ont utilisé des modèles d'apprentissage profond (intelligence artificielle) pour identifier les caractéristiques des séquences qui influencent la formation et l'activité de la chromatine sur ces molécules d'ADN intégrées.
Leur analyse a tout d’abord révélé que quelle que soit sa composition, l’ADN bactérien introduit dans le génome eucaryote de la levure adopte une organisation similaire à celle de l’ADN de l’hôte sous forme de chromatine, c’est-à-dire que l’ADN est enroulé et compacté autour de protéines ayant une forte affinité pour l’ADN (les histones) pour former des nucléosomes. Cependant, la composition de ces séquences va avoir un impact important sur leur activité transcriptionnelle. Ainsi, des séquences exogènes, bactériennes ou autres, dont la proportion de GC se rapproche de celle de la séquence de la levure, sont activement transcrites et se mêlent au sein de l'espace nucléaire aux chromosomes de levure qui sont également activement transcrits. La transcription de ces séquences suit majoritairement l’orientation des gènes bactériens, suggérant un biais directionnel intrinsèque de la RNA polymérase, présent avant la divergence entre eucaryotes et procaryotes. Ce biais pourrait jouer un rôle dans l’adoption des séquences au cours des transferts horizontaux d’ADN. En revanche, les chromosomes pauvres en GC et donc riches en bases AT (adénine-thymine) restent inactives transcriptionnellement. Ces chromosomes adoptent alors une structure très particulière, puisqu’ils sont mis à l’écart des chromosomes de levure actifs et forment un compartiment globulaire à la périphérie du noyau. Ce phénomène conduit donc à la partition du génome hybride en deux compartiments : un actif et un inactif, spatialement distincts au sein du noyau. Les travaux montrent par ailleurs que cette compartimentation disparaît lorsque la transcription est bloquée sur l’ensemble du génome de levure, ce qui suggère que des contraintes mécaniques ou des modifications biochimiques de l'environnement autour des gènes actifs conduisent à la ségrégation des séquences actives et inactives. Cette organisation évoque, sous certains aspects, la compartimentation des génomes de métazoaires (comme les mammifères) en euchromatine (régions actives) et hétérochromatine (régions inactives). Une différence majeure est que, chez ces espèces, cette partition est régulée par la formation d’hétérochromatine canonique sur les séquences inactives, via H3K9me3, un mécanisme absent chez S. cerevisiae. Ainsi, il est possible que la partition observée chez S. cerevisiae, seulement dépendante de l’activité transcriptionnelle, représente un processus spontané permettant notamment de traiter le devenir de séquences exogènes, qui aurait par la suite été complexifié par apparition de mécanismes plus spécialisés. Au cours de la mitose et de la ségrégation lors de la division cellulaire, la compartimentation disparaît lorsque la chromatine est compactée en chromosomes mitotiques sous l’effet de boucles formées par une protéine, la cohésine, un phénomène ici aussi similaire à la perte transitoire des compartiments observée chez les métazoaires à ce stade.
Par ailleurs, les scientifiques ont démontré que des modèles d’apprentissage profond (i.e. intelligence artificielle), entraînés sur une partie des chromosomes de levure, permettent de prédire avec précision la composition et l’activité de la chromatine sur de l’ADN étranger en fonction de sa séquence. Cela suggère que le comportement de tout ADN présent dans une cellule hôte suit des règles déterministes basées sur sa séquence. Cette observation pourrait aider à prédire le comportement d'ADN exogène non seulement pendant les événements naturels de transfert de gènes entre espèces, mais aussi dans les projets d'ingénierie de génomique synthétiques.
La formation spontanée de compartiments transcriptionnellement actifs et inactifs dans un génome hôte eucaryote dépourvu de telles structures pourrait permettre de mieux comprendre l'émergence des compartiments chromatiniens au cours de l'évolution. L'intégration de séquences d'ADN exogènes dans le génome de la levure entraîne une organisation chromatinienne dictée par leur composition, influençant ainsi leur capacité à recruter la machinerie transcriptionnelle de l’hôte. Ces propriétés, que l’on peut prédire à partir de la séquence, mettent en évidence les liens fondamentaux entre la composition de la séquence d'ADN, l'organisation de la chromatine et l'architecture nucléaire. Ces caractéristiques dépendantes de la séquence pourraient avoir contribué à l'émergence d'une compartimentation bipartite des génomes au cours de l'évolution, soit en séquestrant le matériel génétique inactif, soit en facilitant l'intégration fonctionnelle des séquences actives, créant ainsi potentiellement des réservoirs de nouveauté génétique.
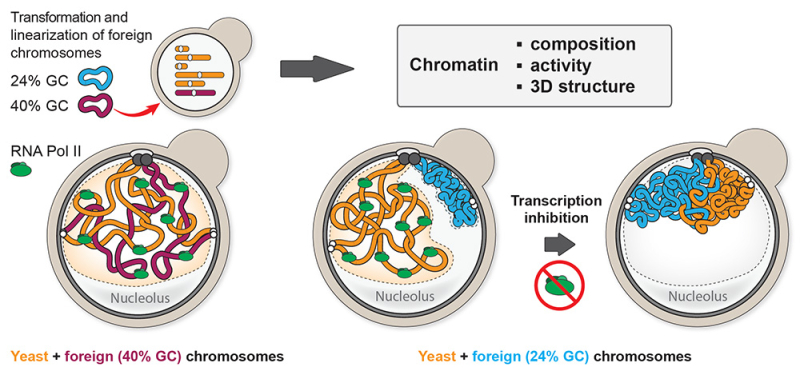
Figure : Différents destins de chromosomes étrangers dans un noyau de levure. Les chromosomes bactériens ou eucaryotes dont la teneur en GC est similaire à celle des séquences de levure (en violet) deviennent activement transcrits et se mélangent aux chromosomes de l'hôte (à gauche). En revanche, les chromosomes étrangers à faible teneur en GC (en bleu) forment un compartiment globulaire distinct, silencieux sur le plan de la transcription, à l'intérieur du noyau (au milieu). Cette ségrégation disparaît lorsque la transcription est inhibée (à droite), mais est indépendante de la formation d'hétérochromatine canonique médiée par le complexe SIR.
En savoir plus : Léa Meneu et al. Sequence-dependent activity and compartmentalization of foreign DNA in a eukaryotic nucleus. Science387, eadm9466(2025). DOI:10.1126/science.adm9466
Dans un article publié dans la revue Nature Structural and Molecular Biology, ...
Génétique des génomes (CNRS/Institut Pasteur)
Institut Pasteur
28 rue du Dr. Roux
75015 Paris
Structure et Instabilité des Génomes - StrInG (CNRS/Inserm/MnHn)
43 Rue Cuvier
75005 Paris