
MethyLasso : un nouvel outil pour analyser les données de méthylation de l’ADN
La méthylation de l’ADN est une modification épigénétique permettant de réprimer l’expression des gènes. Dans un article publié dans Nucleic Acids Research, des scientifiques décrivent une nouvelle méthode bio-informatique permettant d’analyser les données de méthylation de l’ADN à l’échelle du génome. Cette approche s’avère utile pour mieux comprendre les changements de méthylation de l’ADN induits dans différentes conditions physiologiques ou pathologiques.
Méthylation de l’ADN, une clé de la régulation génétique
La méthylation de l’ADN est une modification épigénétique qui modifie les cytosines composant l’ADN en y ajoutant un groupement méthyle. La majorité des cytosines sont normalement méthylées, ce qui permet de conserver le génome réprimé. Seules certaines régions régulatrices sont activées de manière spécifique afin de permettre l’activation de gènes important à la fonction de chaque cellule. Cette activation est induite par la liaison au génome de protéines appelées facteurs de transcription et engendre une perte de méthylation de l’ADN au niveau de ces régions génomiques.
Les approches de séquençage à haut débit permettent maintenant de cartographier les niveaux de méthylation de l’ADN sur génome entier. Cependant, le développement de méthodes bio-informatiques est encore nécessaire afin d’analyser les données et d’en extraire le maximum d’information possible.
MethyLasso, une méthode innovante pour analyser les méthylations de l’ADN
Dans un article publié dans la revue Nucleic Acids Research, des scientifiques ont développé une méthode innovante appelée MethyLasso qui permet une analyse fine des données de méthylation de l’ADN. Cette approche effectue tout d’abord une segmentation des données afin d’identifier des régions du génome qui sont déméthylées et qui correspondent aux régions régulatrices actives liées par des facteurs de transcription. MethyLasso permet aussi d’identifier des régions avec des niveaux de méthylation hétérogènes qui correspondent notamment à de large régions inactives dans les données provenant de cancers.
Enfin, MethyLasso permet aussi d’identifier des régions différentiellement méthylées lors de comparaisons entre deux conditions comme par exemple entre cellules saines et cancéreuses. En effet, une perte de méthylation de l’ADN peut induire à l’activation d’oncogènes alors qu’un gain de méthylation de l’ADN peut induire à l’inactivation de gènes suppresseurs de tumeur.
Le nouvel outil MethyLasso permet donc aux scientifiques étudiant la méthylation de l’ADN de réaliser une analyse fine de ces données afin de mieux caractériser les dérégulations observées dans différentes conditions pathologiques.
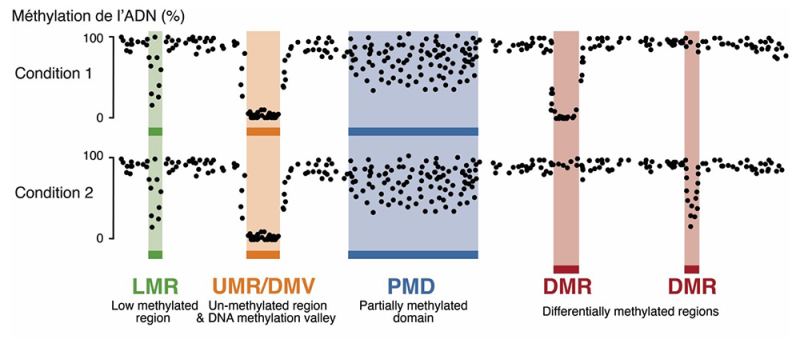
Figure : Chaque point représente une cytosine le long du génome et son niveau de méthylation dans une condition donnée. MethyLasso permet d’identifier des régions du génome avec un faible niveau de méthylation de l’ADN (LMR, UMR et DMV) qui représente les régions régulatrice active du génome, des régions avec un niveau de méthylation de l’ADN très variable (PMD) qui sont présente seulement dans certaines conditions comme par exemple des cellules cancéreuses ainsi que des régions différentiellement méthylés entre deux conditions comme par exemple entre cellules saines et cancéreuses et qui indiques des changements d’activité menant à une dérégulation de l’expression des gènes.
Pour en savoir plus :
MethyLasso: a segmentation approach to analyze DNA methylation patterns and identify differentially methylated regions from whole-genome datasets.
D. Balaramane, Y. G. Spill, M. Weber, A. Bardet.
Nucleic Acids Research, 18 octobre 2024. DOI : 10.1093/nar/gkae880
Contact
Laboratoires
Institut de génétique et de biologie moléculaire et cellulaire - IGBMC (CNRS / Inserm / Université de Strasbourg)
1 rue Laurent Fries,
67404 Illkirch Cedex
Biotechnologie et signalisation cellulaire - BSC (CNRS / Université de Strasbourg)
300 Bd Sébastien Brant,
67412 Illkirch Cedex