
La méthode idéale pour intégrer vos données multi-omiques existe-t-elle ?
Les technologies à haut-débit génèrent des données biologiques omiques en grande quantité, suscitant des défis considérables pour leur analyse et leur intégration. Dans une étude publiée dans la revue Nature Communications, les scientifiques ont évalué en détails neuf approches représentatives de l’état de l’art pour l’extraction concertée des signaux biologiques à partir de données multi-omiques obtenues sur des échantillons de tumeurs. Les résultats, disponibles sous forme de pipeline informatique reproductible, offrent un guide précis pour les utilisateurs et un environnement pouvant accueillir de futurs développements.
Des données moléculaires sont de nos jours produites en masse, suscitant des défis considérables pour leur analyse. Ces données omiques décrivent par exemple l’expression des gènes (transcriptome), l’abondance des protéines (protéome) ou la méthylation de l’ADN (méthylome) provenant d’une population de cellules, voire même, grâce aux progrès techniques, de cellules uniques. Les différentes données omiques permettent de capturer des informations sur les processus biologiques se déroulant à différentes échelles de la cellule. Des outils bioinformatiques spécifiques sont développés pour analyser ces données de manière indépendante. Cependant, une analyse intégrée et simultanée des différentes données omiques est essentielle pour une compréhension fine des mécanismes cellulaires ainsi que de leurs dérégulations pathologiques. Le développement de telles méthodes d'analyses intégratives est un défi majeur en bioinformatique.
Parmi les approches permettant l’analyse intégrative de différentes données omiques, les approches de factorisation de matrices se sont révélées particulièrement efficaces. Chaque jeu de données omiques est représenté sous forme d’une (très grande) matrice. Ces matrices sont ensuite factorisées, c'est-à-dire décomposées en produit de deux matrices, avec la contrainte qu’une des matrices du produit soit commune aux différentes données. Cette technique permet de réduire la dimension des données ainsi que d’en extraire le signal biologique conjoint.
Ces méthodes de factorisation de matrices ont été appliquées à la recherche sur le cancer, où l'intégration des données obtenues à différentes échelles est essentielle pour démêler les sous-types et les mécanismes physiopathologiques sous-jacents. L’objectif est de permettre une meilleure gestion et de meilleurs traitements, dans le cadre de la médecine de précision. Cependant, un grand nombre de méthodes de factorisation de matrices existent, basées sur différentes hypothèses et formulations mathématiques. Il y a aujourd'hui un besoin urgent d’évaluation et de comparaison de ces méthodes, et de directives claires pour aider les utilisateurs à choisir celles les plus adaptées à leur cas d’étude.
Les scientifiques ont sélectionné et décrit neuf approches de factorisation de matrices dédiées à l’intégration de données multi-omiques représentatives de l'état de l'art. Ils ont systématiquement évalué leurs performances par des comparaisons rigoureuses dans différents scénarios pertinents pour la recherche sur le cancer. Les performances des méthodes pour identifier des sous-types de cancers sont évaluées sur des données simulées et des données réelles de l’Atlas du génome du cancer. Les méthodes sont comparées sur leurs capacités à prédire les annotations cliniques et de survie des échantillons, ainsi qu’à classifier des échantillons obtenus sur des cellules uniques.
Les résultats permettent d’identifier la meilleure approche lorsque l’objectif est de classer les échantillons et d’identifier les sous-types, ainsi que l’approche la plus versatile lorsque les objectifs biologiques sont moins précisément définis. Enfin, un notebook Jupyter nommé momix permet de reproduire l’ensemble des résultats. Il peut également être utilisé pour tester les méthodes existantes sur de nouveaux jeux de données, ou pour évaluer les performances de méthodes de nouvelle génération.
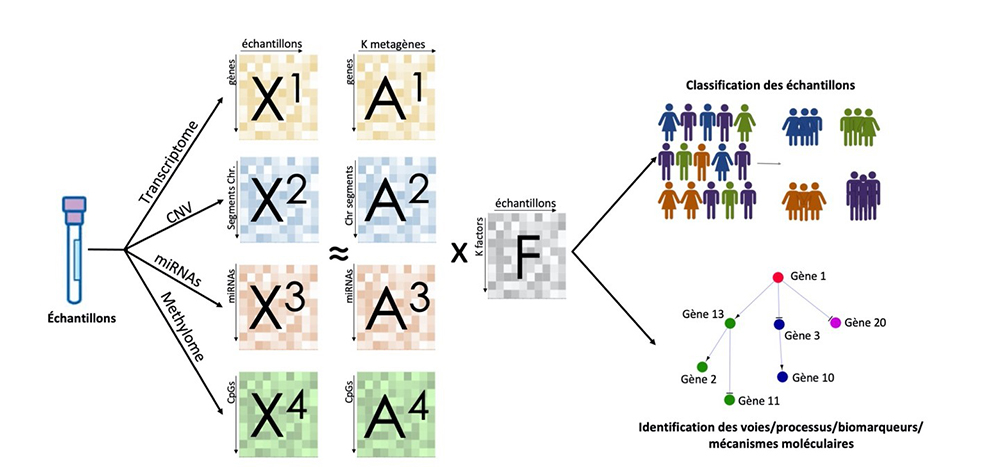
Figure : Workflow d’intégration de données multi-omiques : les différentes données omiques sont factorisées conjointement ; les facteurs obtenus permettent de classer les échantillons et d’identifier les processus biologiques d’intérêt
Pour en savoir plus :
Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer.
Cantini L, Zakeri P, Hernandez C, Naldi A, Thieffry D, Remy E, Baudot A.
Nat Commun. 2021 Jan 5;12(1):124. doi: 10.1038/s41467-020-20430-7.
Contact
Laboratoire
Institut de Biologie de l'École Normale Supérieure (IBENS) - (CNRS, Inserm, Université PSL)
75005, Paris, France
Centre de Génétique Médicale de Marseille - (Inserm, Aix-Marseille Université)
27 Bd Jean Moulin
13385 Marseille Cedex 5