
Empêcher la dégradation du suppresseur de tumeur VHL
L’inactivation du gène codant le suppresseur de tumeur von Hippel-Lindau (VHL) provoque un syndrome héréditaire favorisant l’apparition de tumeurs dans divers organes. Certaines mutations de VHL peuvent affecter le repliement de la protéine pVHL et donc sa fonctionnalité. Dans cet article, publié dans la revue PLos Genetics, les scientifiques ont identifié un complexe chaperon appelé préfoldine qui se lie à VHL. En absence de préfoldine, pVHL mal structurée peut s’agréger et/ou être dégradée. Le niveau d’expression de PFDN3, un composant du complexe préfoldine, est corrélé à la malignité de tumeurs rénales.
Des mutations dans le gène humain von Hippel Lindau (VHL) sont responsables de la maladie de VHL, un syndrome de cancer autosomique dominant qui prédispose à diverses tumeurs bénignes et malignes, y compris le carcinome rénal à cellules claires (ccRCC), l'hémangioblastome (tumeur du système nerveux central) ou le phéochromocytome (tumeur surrénalienne). De plus, le gène VHL est muté ou absent dans 80 % des cas de ccRCC sporadiques, le sous-type le plus courant de cancer du rein.
Le gène VHL code une petite protéine de 213 acides aminés, pVHL, dont les différentes fonctions lui confèrent une activité de suppresseur de tumeur. .
En particulier, la protéine pVHL appartient à un complexe VBC de type ubiquitine-ligase qui est responsable de la destruction de cibles protéiques impliquées dans la croissance et la vascularisation de tumeurs solides. Le substrat le plus connu du complexe VBC est HIFa, qui contrôle la réponse des cellules aux niveaux d'oxygène.
Environ un tiers des mutations qui inactivent pVHL sont des mutations faux-sens produisant une protéine complète mais non fonctionnelle. Certaines de ces mutations se situent en dehors des domaines fonctionnels connus de pVHL et leur impact sur sa fonction reste à élucider. Les chercheurs font l'hypothèse que certaines de ces mutations affectent le repliement optimal de pVHL et favorisent sa dégradation.
Des protéines appelées « chaperons moléculaires » facilitent le repliement et empêchent l'agrégation des polypeptides dans l'environnement dense du cytoplasme en protégeant les zones hydrophobes à la surface des protéines non repliées. Les protéines mal repliées sont détectées par des voies de « contrôle qualité des protéines » qui les ciblent vers la machinerie de dégradation des protéines afin d'empêcher la formation de protéines non fonctionnelles ou d'agrégats cytotoxiques. Le complexe préfoldine est un co-chaperon hexamérique conservé des archae aux eucaryotes. Il joue un rôle important dans le système de contrôle qualité des protéines. Le complexe préfoldine eucaryote est constitué de deux sous-unités alpha (PFDN3, PFDN5) et de quatre sous-unités béta (PFDN1, PFDN2, PFDN4 et PFDN6). Une fois le complexe préfoldine assemblé, sa structure rappelle une méduse avec des « tentacules » (les extrémités des sous-unités) permettant de capter les protéines nouvellement synthétisées non repliées. Le complexe préfoldine transfère ces protéines (par exemple l'actine et la tubuline) non repliées au complexe chaperonine CCT/TRiC pour aider à leur repliement et empêcher leur agrégation.
Chez la levure Schizosaccharomyces pombe, seules deux sous-unités du complexe préfoldine (Pfd3 et Pfd5) ont été caractérisées jusqu'à présent. Les chercheurs avaient récemment observé que Pfd3 contrôlait le niveau d'expression de la protéine pVHL humaine exprimée de manière exogène chez S. pombe. Dans ce travail, ils montrent que pVHL s'associe aux 6 sous-unités du complexe préfoldine. Chez la levure S. pombe, la fonction de chaperon du complexe préfoldine est conservée pour la tubuline et des mutants de toutes les sous-unités de ce complexe présentent des défauts d’assemblage des microtubules. En utilisant un système d'expression conditionnelle, les chercheurs montrent que l'ensemble du complexe préfoldine protège pVHL de l'agrégation, améliore sa solubilité et son repliement. L'expression de la sous-unité PFDN3 humaine chez un mutant délété du gène (pfd3D) de levure restaure la solubilité de pVHL, ce que ne fait pas une PFDN3 inactive (déficiente pour la fonction chaperon). Cela indique que cette fonction chaperon est conservée de la levure à l’homme. Dans les cellules humaines, la diminution de l’expression de l'ensemble du complexe préfoldine par « RNA silencing » affecte aussi l’organisation du réseau de microtubules et réduit les niveaux d'expression de pVHL. La région codée par VHL à la jonction de l’exon2 et de l’exon3, riche en acides aminés hydrophobes, est déterminante pour la fixation du complexe préfoldine. Dans les bases de données TCGA (« The Cancer Genome Atlas ») pour le ccRCC, un faible niveau d'expression de PFDN3 est corrélé à une survie significativement réduite chez les patients porteurs de certaines mutations de VHL.
Ces données suggèrent que l'interaction du complexe préfoldine avec pVHL est une étape clé dans le processus de repliement de pVHL pour atteindre une conformation native qui lui permette d’assurer ses fonctions cellulaires. Ainsi, la fonction du complexe préfoldine peut avoir un impact sur l’homéostasie protéique de pVHL et cela pourrait influencer la progression des maladies associées à VHL chez les patients.
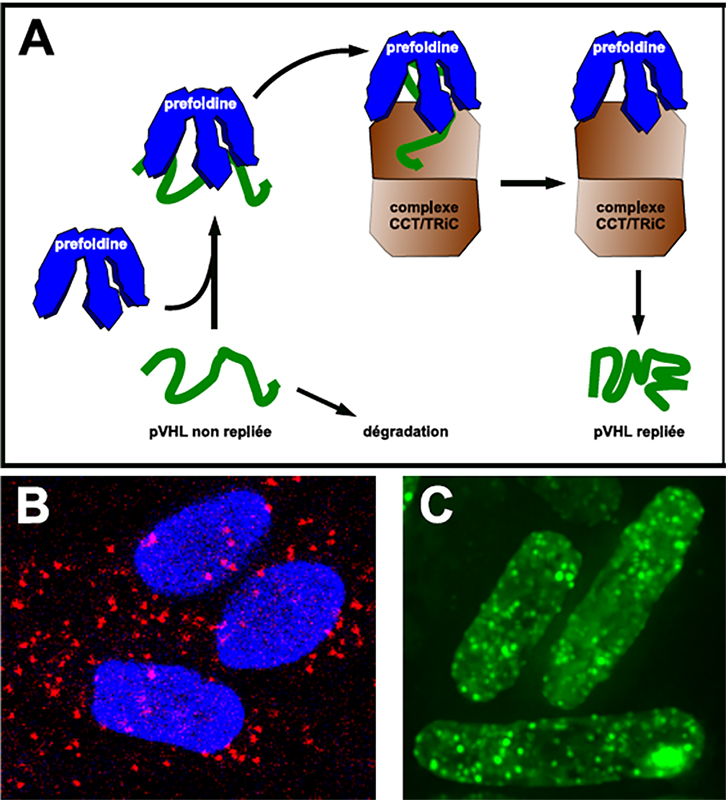
Figure : A) Le complexe préfoldine (bleu) fixe la protéine suppresseur de tumeur pVHL (vert) afin de permettre son repliement et de la rendre fonctionnelle via un autre complexe, le complexe CCT/TRiC. Sans la fixation au complexe préfoldine, la protéine pVHL non repliée est dégradée par les cellules. B) Le complexe préfoldine et la protéine pVHL sont co-localisés (points rouges) dans des cellules rénales humaines en culture (bleu, noyaux des cellules). C) La protéine pVHL fluorescente est mal repliée dans des cellules de levures pfd3D mutantes pour une sous-unité du complexe préfoldine et forme des agrégats.
Pour en savoir plus :
The prefoldin complex stabilizes the von Hippel-Lindau protein against aggregation and degradation.
Chesnel F, Couturier A, Alusse A, Gagné JP, Poirier GG, Jean D, Boisvert FM, Hascoet P, Paillard L, Arlot-Bonnemains Y, Le Goff X.
PLoS Genet. 2020 Nov 2;16(11):e1009183. doi: 10.1371/journal.pgen.1009183
Contact
Laboratoire
Institut de Génétique et Développement de Rennes (IGDR) - (CNRS/Université de Rennes 1)
Faculté de Médecine
2 avenue du Pr Léon Bernard
CS 34317
F-35043 Rennes Cedex
France