
Analyses phylogénétiques du SARS-CoV-2 : forces, limites et surinterprétations
Le 28 Avril paraissait dans la revue PNAS un article sur l’analyse par réseau phylogénétique de génomes du SARS-CoV-2. Cet article proposait une histoire de la pandémie et l’existence de trois sous-types, affectant différentes populations. Cet article était repris par la presse grand public, mais la méthode utilisée avait de nombreuses faiblesses. Dans une réponse collective publiée dans la même revue le 7 Mai, les scientifiques rappellent la difficulté d’enraciner la pandémie avec les données actuelles et la limite des approches phylogéographiques face aux biais d’échantillonnage. Plus encore, ils soulignent le danger de surinterpréter de tels résultats qui reposent sur de nombreuses hypothèses et des données limitées.
L’article de Forster et al. (PNAS, 28 Avril 2020) utilisait une méthode de réseaux phylogénétiques, très largement répandues en génétique humaine car elle permet d’intégrer la recombinaison, mais très rarement en épidémiologie moléculaire. Les virus recombinent, par exemple le VIH et les coronavirus en général, mais à l’échelle du SARS-CoV-2 dont l’apparition date d’environ 6 mois, on n’a pas observé de recombinaison chez l’hôte humain. Les arbres phylogénétiques, plutôt que les réseaux, paraissent donc la bonne approche. Une autre spécificité de la méthode utilisée par Forster et al. est que c’est une méthode exploratoire donnant une représentation visuelle des données, plutôt qu’un méthode inférentielle permettant de tester une hypothèse et de la retenir ou la rejeter avec un certain niveau de confiance statistique. L’interprétation des résultats de la méthode employée par Forster et al. est donc avant tout visuelle et on manque d’indicateur pour en vérifier la robustesse. Une autre limite de cette étude était le très petit nombre de génomes utilisés (160), alors que le jour de la parution de l’article il y en avait 6000 disponibles. A tout le moins, il aurait fallu refaire l’analyse avec des échantillons différents et vérifier la stabilité des résultats. Pour autant l’article a été publié, avec des interprétations nombreuses, notamment l’existence de trois sous-types : A « originel, issu de la chauve-souris et du pangolin », B « issu de A, adapté aux populations asiatiques », C « issu de B et affectant l’Europe et l’Amérique ». Et ces interprétations ont été largement reprises dans les jours qui ont suivi par la presse grand public, jusqu’à déclencher un Tweet de Donald Trump. Dans une période moins troublée, cet article aurait été révisé et ses interprétations revues à la baisse.
Cette surinterprétation d’une étude modeste (160 génomes sur des milliers disponibles) a déclenché une réaction de la communauté internationale. Ainsi, un collectif de près de 40 chercheurs internationaux a posté une lettre, publiée dans la même revue une dizaine de jours après la publication originale (deux autres lettres ont été publiées le même jour sur le même sujet). Dans cette lettre sont relevées deux faiblesses majeures de l’étude de Forster et al. :
1°) La difficulté d’enraciner l’histoire évolutive de cette pandémie en se basant sur les séquences aujourd’hui disponibles. En effet, les séquences humaines ont très peu évolué depuis le mois de décembre 2019 et les premiers prélèvements. Le rythme est d’une à deux mutations par mois pour un génome à ARN d’environ 30 000 bases. Les séquences les plus éloignées observées aujourd’hui présentent au plus une cinquantaine de mutations de différence. A l’inverse le génome de virus animal le plus proche, issu de la chauve-souris, a environ 1 200 différences avec le virus humain. En raison de l’aspect aléatoire des mutations, on ne peut donc pas dire que telle ou telle séquence humaine est significativement plus proche de la séquence de chauve-souris, et constitue LA séquence ancestrale, comme l’ont affirmé Forster et al. A cette difficulté s’ajoute le fait que la séquence la plus fréquente trouvée en Chine en décembre, a aussi été trouvée parfaitement conservée à Taiwan, au Japon, aux USA, au Royaume Uni, etc., et ce jusque récemment. Il est donc difficile d’enraciner la pandémie et de lui donner une origine géographique. On peut sans doute le faire en se basant sur l’histoire des faits et les cas documentés de transmission, mais il faut s’entourer de beaucoup de précautions.
2°) Les biais d’échantillonnage. Dans l’étude de Forster et al. il y avait beaucoup de séquences chinoises, mais, par exemple, peu de séquences italiennes. Aujourd’hui près de la moitié des séquences publics viennent du Royaume-Uni. Dans un cas comme dans l’autre, les représentations sont biaisées, et les méthodes de phylogéographie sont sensibles à ces biais. Elles fonctionnent par reconstruction ancestrale : typiquement la racine d’un sous-arbre dont la grande majorité des feuilles vient d’un pays donné sera attribuée à ce pays. Et ces reconstructions ancestrales sont inévitablement sensibles aux biais de représentation. Si aujourd’hui on fait une analyse naïve avec la moitié des séquences venant du Royaume-Uni, on conclura avec « certitude » que la pandémie vient de là. Il n’est pas facile de compenser ces biais, mais une étude de robustesse permet de vérifier que les résultats ne sont pas excessivement sensibles aux variations des représentations géographiques.
Les analyses phylogénétiques apportent de nombreuses informations en matière d’épidémiologie. Pour le SARS-CoV-2, elles ont par exemple permis de trouver les génomes de virus animaux les plus proches, d’écarter l’hypothèse d’une création humaine en laboratoire, et de dessiner à gros traits les flux entre pays et continents à la surface du globe. Mais elles se basent sur des hypothèses (par exemple la prévalence géographique), et des données incomplètes (par exemple la rareté des séquences italiennes). Il convient de garder en mémoire ces limites, de les expliciter dans les articles scientifiques, et de les rappeler aux journalistes de la presse grand public. En ceci elles ne se distinguent pas de bien d’autres approches scientifiques, mais les précautions sont essentielles sur des sujets sensibles comme la pandémie du COVID-19.
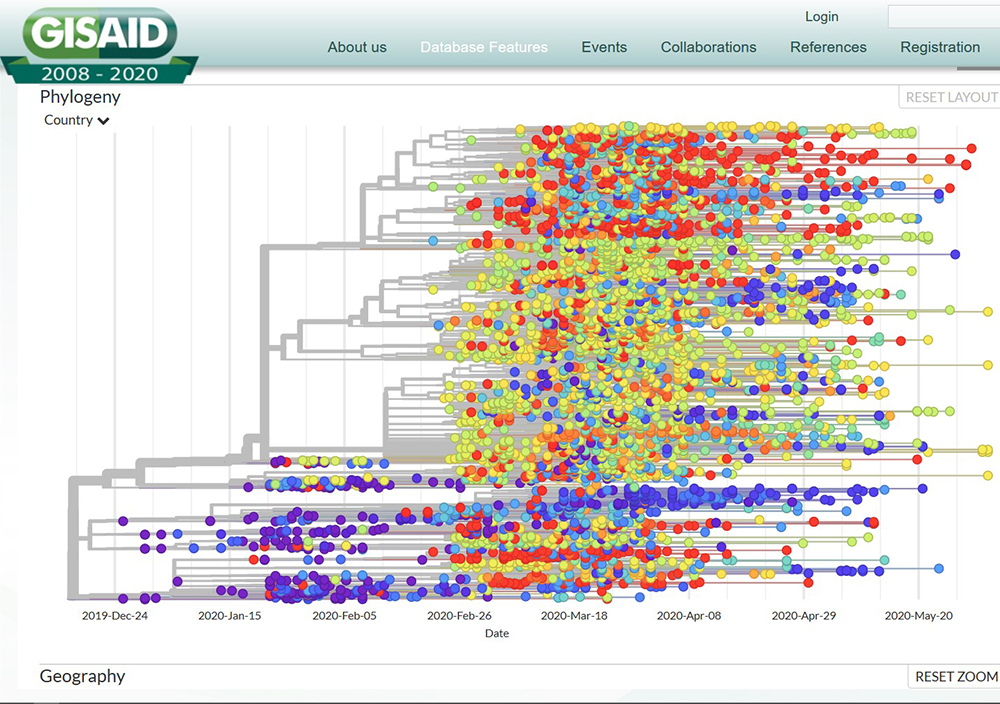
Figure : Phylogéographie proposée par NEXSTRAIN et le GISAID le 7 Juin, portant sur ~4500 génomes échantillonnés depuis fin décembre 2019. Chaque point coloré correspond à un génome. Le violet correspond à la Chine, le rouge aux USA, le vert aux Pays-Bas, le jaune au Royaume Uni, l’orange au Brésil, etc. On voit ici la complexité de l’histoire de la pandémie et la multiplicité des introductions dans n’importe quel pays (USA par exemple, trouvé dans tout l’arbre). Même si on peut dégager des grandes tendances (par exemple l’intrication des épidémies au Royaume Uni, au Pays Bas et au Brésil), il faut prendre garde à ne pas simplifier à l’excès et se souvenir des hypothèses ayant conduit à une telle représentation.
Pour en savoir plus :
Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable.
Mavian C, Pond SK, Marini S, Magalis BR, Vandamme AM, Dellicour S, Scarpino SV, Houldcroft C, Villabona-Arenas J, Paisie TK, Trovão NS, Boucher C, Zhang Y, Scheuermann RH, Gascuel O, Lam TT, Suchard MA, Abecasis A, Wilkinson E, de Oliveira T, Bento AI, Schmidt HA, Martin D, Hadfield J, Faria N, Grubaugh ND, Neher RA, Baele G, Lemey P, Stadler T, Albert J, Crandall KA, Leitner T, Stamatakis A, Prosperi M, Salemi M.
Proc Natl Acad Sci U S A. 2020 May 7:202007295. doi: 10.1073/pnas.2007295117. Online ahead of print. PMID: 32381734
Contact
Laboratoire
Centre de Bioinformatique, Biostatistique et Biologie Intégrative (C3BI) - (Institut Pasteur/CNRS)
28 rue du Docteur Roux, Paris-75015